Molecular biology
How to determine a protein’s shape
Only a third of known protein structures are human
ABOUT 120,000 types of protein molecule have yielded up their structures to science. That sounds a lot, but it isn’t. The techniques, such as X-ray crystallography and nuclear-magnetic resonance (NMR), which are used to elucidate such structures do not work on all proteins. Some types are hard to produce or purify in the volumes required. Others do not seem to crystallise at all—a prerequisite for probing them with X-rays. As a consequence, those structures that have been determined include representatives of less than a third of the 16,000 known protein families. Researchers can build reasonable computer models for around another third, because the structures of these resemble ones already known. For the remainder, however, there is nothing to go on.
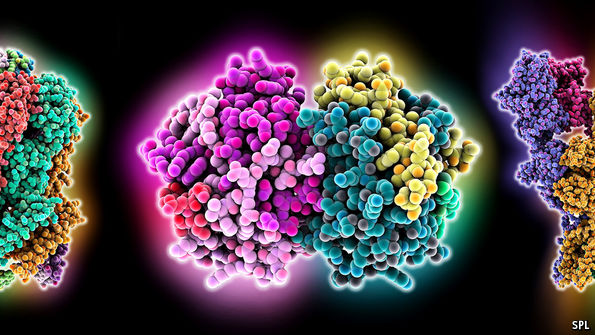
In addition to this lack of information about protein families, there is a lack of information about those from the species of most interest to researchers: Homo sapiens. Only a quarter of known protein structures are human. A majority of the rest come from bacteria. This paucity is a problem, for in proteins form and function are intimately related. A protein is a chain of smaller molecules, called amino acids, that is often hundreds or thousands of links long. By a process not well understood, this chain folds up, after it has been made, into a specific and complex three-dimensional shape. That shape determines what the protein does: acting as a channel, say, to admit a chemical into a cell; or as an enzyme to accelerate a chemical reaction; or as a receptor, to receive chemical signals and pass them on to a cell’s molecular machinery. (Models of all three, in that order, are shown above.)
Almost all drugs work by binding to a particular protein in a particular place, thereby altering or disabling that protein’s function. Designing new drugs is easier if binding sites can be identified in advance. But that means knowing the protein’s structure. To be able to predict this from the order of the amino acids in the chain would thus be of enormous value. That is a hard task, but it is starting to be cracked.
Chain gang
Even a small protein can fold up into tens of thousands of shapes that are more or less stable.
According to Dr Baker, a chain a mere 70 amino acids long—a tiddler in biological terms—has to be folded virtually inside a computer about 100,000 times in order to cover all the possibilities and thus find the optimum. Since it takes a standard microprocessor ten minutes to do the computations needed for a single one of these virtual foldings, even for a protein this small, the project has, for more than a decade, relied on cadging processing power from thousands of privately owned PCs.
Volunteers download a version of Dr Baker’s program, called rosetta@home, that runs in the background when a computer is otherwise idle.
This “citizen science” has helped a lot. But the real breakthrough, which led to those 672 novel structures, is a shortcut known as protein-contact prediction. This relies on the observation that chain-folding patterns seen in nature bring certain pairs of amino acids close together predictably enough for the fact to be used in the virtual-folding process.
An amino acid has four arms, each connected to a central carbon atom. Two arms are the amine group and the acid group that give the molecule its name. Protein chains form because amine groups and acid groups like to react together and link up. The third is a single hydrogen atom. But the fourth can be any combination of atoms able to bond with the central carbon atom. It is this fourth arm, called the side chain, which gives each type of amino acid its individual characteristics.
One common protein-contact prediction is that, if the side chain of one member of a pair of amino acids brought close together by folding is long, then that of the other member will be short, and vice versa. In other words, the sum of the two lengths is constant. If you have but a single protein sequence available, knowing this is not much use. Recent developments in genomics, however, mean that the DNA sequences of lots of different species are now available.
Since DNA encodes the amino-acid sequences of an organism’s proteins, the composition of those species’ proteins is now known, too. That means slightly different versions, from related species, of what is essentially the same protein can be compared. The latest version of Rosetta does so, looking for co-variation (eg, in this case, two places along the length of the proteins’ chains where a shortening of an amino acid’s side chain in one is always accompanied by a lengthening of it in the other). In this way, it can identify parts of the folded structure that are close together.
Though it is still early days, the method seems to work. None of the 614 structures Dr Baker modelled most recently has yet been elucidated by crystallography or NMR, but six of the previous 58 have. In each case the prediction closely matched reality. Moreover, when used to “hindcast” the shapes of 81 proteins with known structures, the protein-contact-prediction version of Rosetta got them all right.
There is a limitation, though. Of the genomes well-enough known to use for this trick, 88,000 belong to bacteria, the most speciose type of life on Earth. Only 4,000 belong to eukaryotes—the branch of life, made of complex cells, which includes plants, fungi and animals. There are, then, not yet enough relatives of human beings in the mix to look for the co-variation Dr Baker’s method relies on.
Others think they have an answer to that problem. They are trying to extend protein-contact prediction to look for relationships between more than two amino acids in a chain. This would reduce the number of related proteins needed to draw structural inferences and might thus bring human proteins within range of the technique. But to do so, you need a different computational approach.
Those attempting it are testing out the branch of artificial intelligence known as deep learning.
Linking the links
Jianlin Cheng, of the University of Missouri, in Columbia, who was one of the first to apply deep learning in this way, says such programs should be able to spot correlations between three, four or more amino acids, and thus need fewer related proteins to predict structures.
Jinbo Xu, of the Toyota Technological Institute in Chicago, claims to have achieved this already. He and his colleagues published their method in PLOS Computational Biology, in January, and it is now being tested.
If the deep-learning approach to protein folding lives up to its promise, the number of known protein structures should multiply rapidly. More importantly, so should the number that belong to human proteins. That will be of immediate value to drugmakers. It will also help biologists understand better the fundamental workings of cells—and thus what, at a molecular level, it truly means to be alive.
0 comments:
Publicar un comentario